Maladie de Wilson : Causes, Symptômes, Diagnostic et Traitement
La maladie de Wilson est une maladie génétique rare mais grave, qui provoque une accumulation excessive de cuivre dans l’organisme, en particulier dans le foie, le cerveau et les yeux. Souvent diagnostiquée à l’adolescence ou chez le jeune adulte, elle peut entraîner de graves complications comme une cirrhose, une insuffisance hépatique ou des troubles neurologiques si elle n’est pas prise en charge à temps.
Bien que méconnue du grand public, la maladie de Wilson est aujourd’hui mieux dépistée grâce aux analyses biologiques, aux examens ophtalmologiques et aux tests génétiques. Une prise en charge précoce et adaptée permet de contrôler l’accumulation du cuivre et d’offrir aux patients une qualité de vie satisfaisante.
👉 Dans cet article, nous allons explorer en détail les causes, symptômes, méthodes de diagnostic et traitements disponibles pour mieux comprendre cette pathologie complexe.
Table of Contents
Définition et généralités
La maladie de Wilson est une maladie génétique héréditaire rare (1 cas sur 30 000 environ) due à une anomalie du gène ATP7B, situé sur le chromosome 13. Cette mutation empêche le corps d’éliminer correctement le cuivre par la bile.
Normalement, une partie du cuivre apporté par l’alimentation est utilisée par l’organisme (enzymes, métabolisme) et l’excédent est éliminé via le foie. Chez les patients atteints de la maladie de Wilson, ce mécanisme est perturbé :
- Le cuivre s’accumule progressivement dans le foie, provoquant une hépatite chronique, une cirrhose ou une insuffisance hépatique.
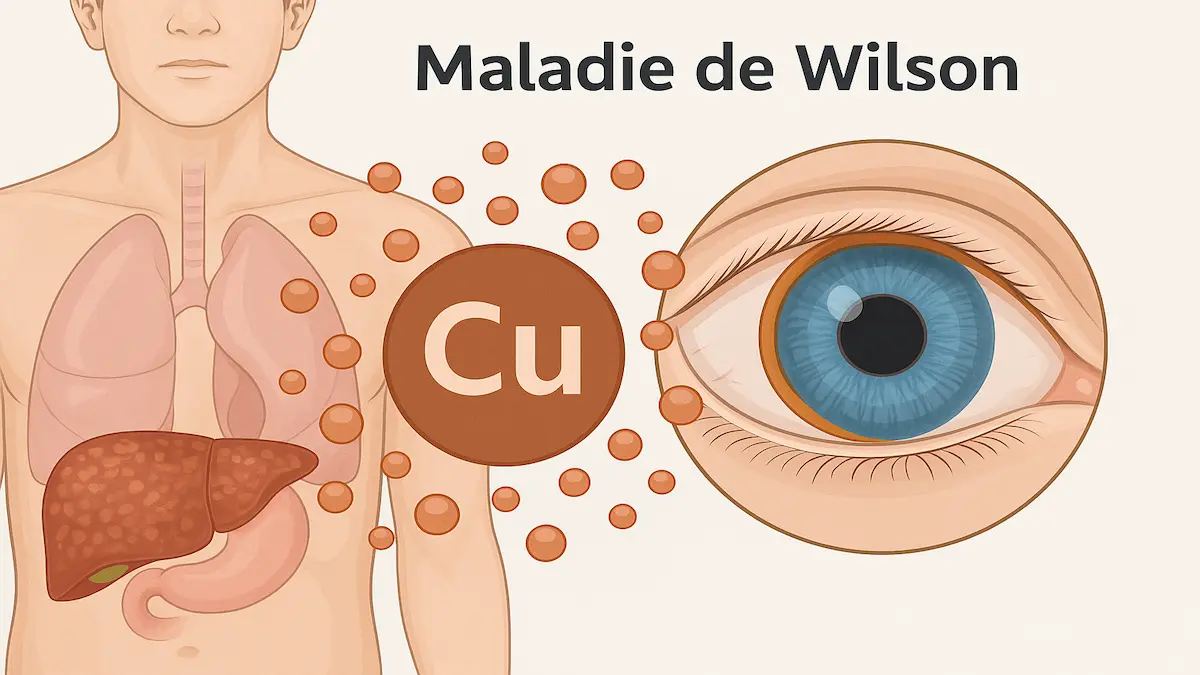
- L’excès de cuivre se dépose ensuite dans le cerveau (atteintes neurologiques et psychiatriques) et les yeux (anneau de Kayser-Fleischer).
La maladie de Wilson est transmise sur un mode autosomique récessif : cela signifie que pour développer la maladie, une personne doit hériter de deux copies mutées du gène (une de chaque parent). Les porteurs d’une seule mutation ne développent pas la maladie mais peuvent la transmettre à leurs enfants.
Bien que rare, cette pathologie est potentiellement mortelle sans traitement, mais une prise en charge précoce permet de contrôler l’évolution et d’améliorer considérablement la qualité de vie des patients.
Causes et mécanismes
La maladie de Wilson est causée par une anomalie du gène ATP7B, qui code une protéine essentielle à l’élimination du cuivre par la bile. Cette mutation entraîne un dysfonctionnement du métabolisme du cuivre :
Mutation génétique
- Le gène ATP7B est situé sur le chromosome 13.
- Sa mutation empêche l’organisme de transporter et d’excréter correctement le cuivre.
- Plus de 500 mutations différentes de ce gène ont été identifiées dans le monde, expliquant la variabilité des symptômes selon les patients.
Transmission autosomique récessive
- Un patient doit hériter de deux copies mutées du gène (une de chaque parent) pour développer la maladie.
- Les individus porteurs d’une seule mutation sont asymptomatiques mais transmettent potentiellement le gène défectueux.
- Le risque pour un enfant d’avoir la maladie est de 25 % si les deux parents sont porteurs.
Conséquences biologiques
- Le cuivre ne peut plus être éliminé efficacement par la bile.
- Il s’accumule dans le foie, entraînant inflammation et fibrose progressive (cirrhose).
- L’excès de cuivre se diffuse dans le sang et se dépose dans d’autres organes :
- Cerveau → troubles neurologiques et psychiatriques.
- Cornée → apparition de l’anneau brun-verdâtre de Kayser-Fleischer.
- Reins et autres tissus → anomalies métaboliques diverses.
👉 Ce mécanisme explique pourquoi la maladie de Wilson est à la fois une maladie hépatique et une maladie systémique.
Symptômes de la maladie de Wilson
La maladie de Wilson se manifeste de façon très variable, ce qui rend parfois le diagnostic difficile. Les premiers signes apparaissent généralement entre 5 et 35 ans, mais des cas plus précoces ou plus tardifs existent.
Atteinte hépatique
- Hépatite chronique : fatigue persistante, élévation des transaminases.
- Cirrhose : ascite, varices œsophagiennes, ictère (jaunisse).
- Insuffisance hépatique fulminante : évolution brutale, urgence vitale.
Atteinte neurologique
- Tremblements : fins ou de grande amplitude.
- Troubles moteurs : rigidité musculaire, dystonie, démarche instable.
- Troubles de la parole et de la déglutition : difficultés à articuler, troubles de la coordination.
Atteinte psychiatrique
- Changements de comportement, irritabilité.
- Dépression ou troubles anxieux.
- Baisse des performances scolaires ou professionnelles.
- Dans certains cas : confusion, désorientation.
Atteinte oculaire
- Anneau de Kayser-Fleischer : dépôt brun-verdâtre de cuivre dans la cornée, visible à l’examen à la lampe à fente.
- Il est présent dans plus de 95 % des cas neurologiques.
Autres manifestations possibles
- Anomalies rénales (protéinurie).
- Troubles hématologiques (anémie hémolytique).
- Atteintes articulaires (douleurs, arthrites).
👉 Cette diversité de symptômes explique pourquoi la maladie de Wilson est souvent confondue avec d’autres maladies hépatiques ou neurologiques avant que le diagnostic ne soit confirmé.
Diagnostic de la maladie de Wilson
Le diagnostic repose sur un ensemble d’examens biologiques, cliniques et génétiques. Comme les symptômes peuvent mimer d’autres maladies du foie ou du système nerveux, il est essentiel de combiner plusieurs approches.
Analyses biologiques
- Céruloplasmine sérique : généralement basse (< 0,2 g/L) chez les patients atteints.
- Cuivre sérique : paradoxalement bas (car lié à la céruloplasmine), mais le cuivre libre est élevé.
- Cuivre urinaire (24 heures) : augmenté (> 100 µg/24h), reflète l’excès de cuivre circulant.
Examens hépatiques
- Biopsie hépatique : permet de mesurer directement la concentration en cuivre dans le foie (> 250 µg/g de poids sec confirme le diagnostic).
- Peut aussi révéler des lésions de cirrhose ou d’hépatite chronique.
Examen ophtalmologique
- Recherche de l’anneau de Kayser-Fleischer à la lampe à fente.
- Très fréquent en cas d’atteinte neurologique.
Tests génétiques
- Identification de la mutation du gène ATP7B.
- Utiles pour confirmer le diagnostic et réaliser un dépistage familial.
Aides au diagnostic
- Score de Leipzig : intègre céruloplasmine, cuivre urinaire, anneau de Kayser-Fleischer et tests génétiques pour établir une probabilité diagnostique.
👉 Un diagnostic précoce est crucial, car un traitement instauré rapidement peut prévenir les complications graves et améliorer considérablement la qualité de vie.
Complications de la maladie de Wilson
En l’absence de diagnostic et de traitement précoce, la maladie de Wilson entraîne une accumulation progressive de cuivre qui endommage plusieurs organes vitaux. Les complications peuvent être sévères et parfois irréversibles.
Complications hépatiques
- Cirrhose : conséquence la plus fréquente, liée à la destruction progressive du foie.
- Insuffisance hépatique chronique : fatigue intense, troubles de la coagulation, ascite.
- Insuffisance hépatique fulminante : urgence vitale, nécessitant souvent une transplantation.
Complications neurologiques
- Tremblements invalidants, rigidité musculaire et troubles moteurs sévères.
- Dysarthrie et troubles de la déglutition pouvant entraîner des difficultés de communication et de nutrition.
- Risque de handicap moteur et perte d’autonomie si la maladie n’est pas traitée.
Complications psychiatriques
- Troubles dépressifs graves.
- Désorientation, confusion, voire démence dans les formes avancées.
- Retentissement majeur sur la vie sociale et familiale.
Autres complications
- Encéphalopathie hépatique : due à l’insuffisance hépatique décompensée.
- Anémie hémolytique (destruction des globules rouges par excès de cuivre).
- Atteintes rénales (calculs, protéinurie) et articulaires (douleurs chroniques).
👉 Ces complications montrent que la maladie de Wilson n’est pas seulement une maladie du foie, mais une pathologie systémique, qui nécessite un suivi médical rigoureux et précoce.
Traitements de la maladie de Wilson
La maladie de Wilson ne peut pas être guérie, mais elle peut être contrôlée efficacement grâce à un traitement adapté et suivi à vie. L’objectif est de réduire l’excès de cuivre dans l’organisme et de prévenir les complications.
Médicaments chélateurs du cuivre
- D-pénicillamine : traitement de première intention depuis plusieurs décennies. Elle favorise l’élimination du cuivre par les urines.
- Inconvénients : effets secondaires possibles (réactions cutanées, atteintes rénales, déficit en vitamine B6).
- Trientine : alternative mieux tolérée chez certains patients.
Traitement par le zinc
- Le zinc réduit l’absorption intestinale du cuivre.
- Il est utilisé en traitement d’entretien ou chez les patients intolérants aux chélateurs.
- Souvent prescrit à vie, même après amélioration clinique.
Transplantation hépatique
- Indiquée dans les formes graves avec insuffisance hépatique fulminante ou cirrhose décompensée.
- La transplantation corrige à la fois la fonction hépatique et le défaut génétique, car le nouveau foie possède un gène ATP7B fonctionnel.
- Les résultats sont généralement très bons avec une nette amélioration de la survie.
Suivi médical
- Le traitement est à vie et nécessite un suivi régulier (bilan sanguin, cuivre urinaire, imagerie du foie).
- Un suivi neurologique et psychiatrique est également indispensable.
- La compliance thérapeutique est essentielle : un arrêt de traitement expose à une rechute grave.
👉 Grâce à ces traitements, la majorité des patients diagnostiqués précocement peuvent mener une vie quasiment normale.
Prévention et qualité de vie
Même si la maladie de Wilson est une pathologie génétique et donc non évitable, il est possible d’agir sur la prévention des complications et d’améliorer considérablement la qualité de vie des patients grâce à un suivi adapté.
Dépistage familial
- Toute personne ayant un membre de sa famille diagnostiqué doit bénéficier d’un test génétique pour vérifier la présence du gène muté.
- Le dépistage permet d’identifier les porteurs asymptomatiques et de mettre en place un suivi précoce.
Surveillance médicale régulière
- Contrôle des bilan sanguins (céruloplasmine, transaminases, cuivre urinaire).
- Suivi hépatique (échographie, fibroscan) pour dépister cirrhose ou complications.
- Évaluation neurologique et psychiatrique régulière pour ajuster la prise en charge.
Adaptation alimentaire
- Limiter les aliments riches en cuivre : chocolat, fruits secs, crustacés, champignons, foie et abats.
- Privilégier une alimentation équilibrée de type méditerranéen.
- Respecter les recommandations du médecin et/ou du diététicien spécialisé.
Qualité de vie et éducation thérapeutique
- L’observance stricte du traitement est indispensable pour maintenir une vie normale.
- Les patients bien traités peuvent étudier, travailler et avoir une vie sociale et familiale satisfaisante.
- Un accompagnement psychologique ou associatif peut aider à mieux gérer la maladie au quotidien.
👉 Avec un diagnostic précoce, un traitement adapté et un suivi régulier, la maladie de Wilson peut être bien contrôlée, offrant aux patients une espérance de vie proche de la normale.
FAQ – Questions fréquentes sur la maladie de Wilson
La maladie de Wilson est-elle curable ?
Non, il n’existe pas de guérison définitive. Cependant, avec un traitement à vie par chélateurs du cuivre ou zinc, la maladie peut être contrôlée efficacement et les patients peuvent mener une vie quasi normale.
Peut-on vivre longtemps avec la maladie de Wilson ?
Oui. Si le diagnostic est fait précocement et que le traitement est suivi rigoureusement, l’espérance de vie est proche de la normale. En revanche, un diagnostic tardif expose à de graves complications hépatiques et neurologiques.
Quels sont les premiers signes qui doivent alerter ?
Les plus fréquents sont une fatigue persistante, des troubles hépatiques (jaunisse, douleurs abdominales, anomalies biologiques), des tremblements, ou encore des changements de comportement (dépression, irritabilité).
Quels aliments éviter en cas de maladie de Wilson ?
Il est recommandé de limiter les aliments riches en cuivre :
- Chocolat, cacao
- Fruits secs (noix, amandes, noisettes)
- Crustacés et fruits de mer
- Champignons
- Foie et abats
Un diététicien spécialisé peut adapter le régime selon chaque patient.
Comment dépister la maladie de Wilson chez les proches ?
Le test génétique est la méthode la plus fiable. Les frères et sœurs d’un patient doivent être dépistés systématiquement, car ils ont un risque de 25 % d’être atteints. Des bilans sanguins et ophtalmologiques peuvent compléter l’évaluation.
Quel est l’âge moyen du diagnostic ?
La maladie est généralement détectée entre 5 et 35 ans, mais elle peut se manifester plus tôt chez l’enfant ou plus tardivement chez l’adulte.
La maladie de Wilson est-elle fréquente ?
C’est une maladie rare : environ 1 personne sur 30 000 est atteinte dans le monde. Sa rareté contribue au retard diagnostique.
Quels examens confirment le diagnostic ?
- Céruloplasmine sérique basse
- Cuivre urinaire élevé
- Biopsie hépatique avec excès de cuivre
- Examen des yeux (anneau de Kayser-Fleischer)
- Analyse génétique du gène ATP7B
La maladie de Wilson peut-elle provoquer des troubles psychiques ?
Oui, certains patients présentent des troubles psychiatriques : dépression, anxiété, irritabilité, baisse de concentration, voire confusion. Ces symptômes peuvent précéder les atteintes hépatiques.
Peut-on avoir des enfants avec la maladie de Wilson ?
Oui, la grossesse est possible avec un suivi médical adapté. Certains traitements doivent être ajustés, et le risque génétique pour l’enfant doit être expliqué (25 % si le partenaire est porteur du gène).
Conclusion
La maladie de Wilson est une pathologie génétique rare mais potentiellement grave si elle n’est pas diagnostiquée à temps. En provoquant une accumulation anormale de cuivre dans le foie, le cerveau et d’autres organes, elle peut évoluer vers une insuffisance hépatique, des troubles neurologiques sévères et des complications psychiatriques.
Heureusement, les connaissances actuelles permettent aujourd’hui un diagnostic plus précoce grâce aux analyses biologiques, aux examens ophtalmologiques et aux tests génétiques. Lorsqu’elle est détectée tôt, la maladie peut être contrôlée efficacement grâce à un traitement adapté (chélateurs du cuivre, zinc, suivi à vie).
👉 Le rôle du dépistage familial est essentiel : toute personne ayant un proche atteint doit bénéficier d’une évaluation, même en l’absence de symptômes.
👉 Une bonne observance thérapeutique et un suivi régulier permettent aux patients de mener une vie quasi normale, en évitant les complications graves.
En résumé, la maladie de Wilson n’est pas une condamnation : c’est une maladie chronique qui, avec un traitement bien conduit, permet une qualité de vie satisfaisante. La clé reste la prévention par le dépistage, l’éducation thérapeutique et la vigilance médicale.